

Thalassemias are hereditary illnesses that cause anemia that typically begins in childhood due to a decrease in the synthesis of hemoglobin's alpha or beta chains. The severity of alpha thalassemia, which results from deletions in the alpha-globin gene, varies according to the number of deleted alleles; a four-allele deletion is fatal. Point mutations in the beta-globin gene cause beta thalassemia, which is divided into two types according to the zygosity of the mutation: beta-thalassemia major (severe, homozygous) and beta-thalassemia minor (mild, heterozygous). Jaundice and severe anemia, which necessitate lifelong transfusions, are signs of beta thalassemia major. While cohabitation with sickle cell trait can result in more severe manifestations, co-inheritance of alpha-thalassemia may lessen symptoms. https://www.ncbi.nlm.nih.gov/books/NBK545151/
There are three primary types of thalassemia: minor, intermedia, and major. Each has unique symptoms and problems. Fatigue, paleness, and a lower red blood cell count—an average of 9.45 g/dL in younger people—are symptoms of moderate anemia caused by thalassemia minor, or the trait. Patients may be more susceptible to infection, and the significance of being aware of consanguineous marriages is emphasized. In addition to more severe symptoms including iron excess, splenomegaly, and persistent anemia, thalassemia intermedia can cause joint pain and skeletal abnormalities. In order to prevent problems including skeletal deformities and organ damage, thalassemia major, which is characterized by severe anemia and growth delays, requires thorough therapy through blood transfusions and medicines. Chronic anemia, growth retardation, and an elevated risk of infection as a result of weakened immunity are other symptoms. Sadiq, I. Z., Abubakar, F. S., Usman, H. S., Abdullahi, A. D., Ibrahim, B., Kastayal, B. S., Ibrahim, M., & Hassan, H. A. (2024). Thalassemia: Pathophysiology, Diagnosis, and Advances in Treatment. Thalassemia Reports, 14(4), 81-102
One of the studies from Foong, W. C., Chean, K. Y., Rahim, F. F., Goh, A. S., Yeoh, S. L., & Yeoh, A. A. C. (2022). Quality of life and challenges experienced by the surviving adults with transfusion dependent thalassaemia in Malaysia: a cross sectional study. Health and Quality of Life Outcomes, 20(1). explores the quality of life (QOL) and challenges faced by adults with transfusion-dependent thalassaemia (TDT) in Malaysia. It involved a survey of 196 participants, revealing that 45% had comorbidities, with a notable percentage suffering from multiple complications. Despite a mean QOL score above 60 across physical, psychological, social, and environmental domains, many reported significant disruptions to daily life and employment challenges, including a high unemployment rate and low wages among employed individuals. Respondents expressed dissatisfaction with healthcare services and a lack of community support, highlighting the need for further qualitative research to better understand their specific challenges and improve tailored support. Overall, while adults with TDT manage relatively well, persistent life challenges remain.
This observational study reports on the status of thalassaemia in Malaysia using data from the Malaysian Thalassaemia Registry, which compiled information from 110 participating hospitals from 2007 to 2018. The findings indicate that 7,984 out of 8,681 registered patients are alive, predominantly in Sabah (22.72%), with the largest affected age group being 5-24.9 years old (64.45%), and most patients are Malay (63.95%). The most common diagnosis is haemoglobin E/β-thalassaemia (34.37%). A significant portion of patients are receiving regular blood transfusions (56.73%) and chelation therapy (61.72%). Despite improvements in managing severe iron overload, cardiac complications remain the leading cause of death. The registry aids in understanding disease progression and informs health policies to enhance treatment outcomes and quality of life for patients. Mohd Ibrahim, H., Muda, Z., Othman, I. S., Mohamed Unni, M. N., Teh, K. H., Thevarajah, A., Gunasagaran, K., Ong, G. B., Yeoh, S. L., Muhammad Rivai, A., Che Mohd Razali, C. H., Din, N. D., Abdul Latiff, Z., Jamal, R., Mohamad, N., Mohd Ariffin, H., & Alias, H. (2020). Observational study on the current status of thalassaemia in Malaysia: a report from the Malaysian Thalassaemia Registry. BMJ open, 10(6), e037974.
Thalassaemia is a genetic blood disorder affecting the production of haemoglobin, leading to varying degrees of anaemia. In Malaysia, 1 in 20 individuals may be carriers. Thalassaemia Minor is asymptomatic, while Thalassaemia Major leads to severe health issues, including life-threatening anaemia and requires lifelong treatment. Symptoms can appear from 3 to 18 months, and affected children may show signs such as fatigue, pallor, and organ complications. Couples are advised to undergo screening for carrier status, and Prenatal Thalassaemia Testing can detect the condition in unborn babies. Treatment options include blood transfusions, iron removal protocols, and potential bone marrow transplantation.https://www.drganpenang.com/thalassaemia/
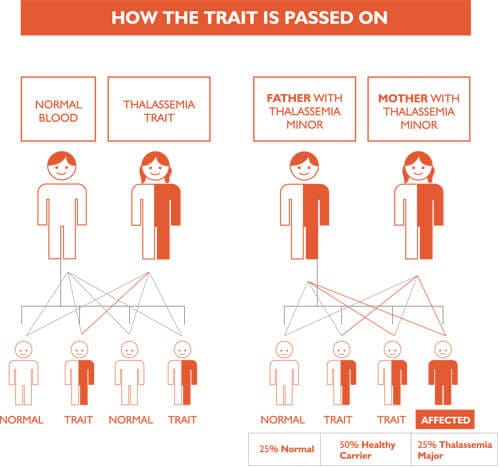
Mirhaa05 (hemirhaphysio@gmail.com) is a content creator under the Newswav Creator programme, where you get to express yourself, be a citizen journalist, and at the same time monetize your content & reach millions of users on Newswav. Log in to creator.newswav.com and become a Newswav Creator now!
The User Content (as defined on Newswav Terms of Use) above including the views expressed and media (pictures, videos, citations etc) were submitted & posted by the author. Newswav is solely an aggregation platform that hosts the User Content. If you have any questions about the content, copyright or other issues of the work, please contact creator@newswav.com.